- Inleiding
- Geneesmiddelen die GGO's bevatten of eruit bestaan
- Besluitvorming
-
Instrumenten voor risicobeoordeling en risicobeheer (nieuwe pagina)
-
Enkele cijfers (nieuwe pagina)
Inleiding
Om in de Europese Unie op de markt gebracht te worden, moet ieder geneesmiddel afkomstig uit de biotechnologie (dus ook geneesmiddelen die GGO's bevatten of eruit bestaan) een toelating krijgen van de Europese Commissie, op advies van EMA (European Medicines Agency). De toegang tot de markt van de Europese Unie voor "GGO" geneesmiddelen wordt door de gecentraliseerde procedure geregeld, zoals gedefinieerd in Verordening 726/2004. Indien de toelating toegekend wordt, is deze meteen geldig voor alle lidstaten van de EU.
De aanvrager van een toelating voor het op de markt brengen van een geneesmiddel dat afkomstig is uit de biotechnologie dient bij EMA een dossier in voor de aanvraag van registratie, dat volgens wetenschappelijke criteria van kwaliteit, veiligheid en doeltreffendheid van het geneesmiddel beoordeeld zal worden. Conform de gecentraliseerde procedure, en afhankelijke van het type geneesmiddel, wordt de beoordeling door EMA uitgevoerd ofwel door het comité voor geneesmiddelen voor menselijk gebruik ("Committee for Medicinal Products for Human Use" - CHMP) door het comité voor geneesmiddelen voor dierlijk gebruik ("Committee for Medicinal Products for Veterinary Use" - CVMP), ofwel door het "Committee for Advanced Therapy" - CAT.
Voor ieder dossier wordt de evaluatie gecoördineerd door een rapporteur en een co-rapporteur, aangeduid door één van de comités. In een eerste instantie wordt een preliminair beoordelingsrapport opgesteld, op basis waarvan vragen aan de kennisgever gesteld kunnen worden. Na diens antwoorden ontvangen te hebben, wordt het finaal beoordelingsrapport ter evaluatie voorgelegd aan het overeenkomend comité van de EMA. Na deze evaluatie beoordeelt het comité de kosten-baten analyse van het gebruik van het geneesmiddel, en finaliseert het diens opinie. Indien deze positief is, wordt die aan de Commissie overgemaakt, die dan de uiteindelijke beslissing neemt.
Geneesmiddelen die GGO's bevatten of eruit bestaan
De geneesmiddelen die GGO's bevatten of eruit bestaan vormen een bijzonder reglementair geval, door de wederzijdse bepalingen aanwezig in Richtlijn 2001/18/EG (art. 12.2) en Verordening (EG) 726/2004 (art. 6.2 en 6.3). In dit geval moet de aanvraag vergezeld zijn van een risicobeoordeling voor het leefmilieu (ERA, environmental risk assessment), zoals aangegeven in Bijlage II van Richtlijn 2001/18/EG. De aanvraag moet dus een volledig technisch dossier bevatten, met de alle informatie gevraagd in Bijlagen III en IV van de Richtlijn.
De ERA is van toepassing op geneesmiddelen die GGO's bevatten of eruit bestaan, maar niet op geneesmiddelen die vanuit GGO's geproduceerd werden. Een voorbeeld van dit laatste is insuline geproduceerd door recombinante bacteriën, of een menselijke bloedfactor, geproduceerd door genetisch gemodificeerde geiten in hun melk.
De ERA wordt geanalyseerd in samenspraak met de structuren opgericht door de Gemeenschap of de Lidstaten, conform Richtlijn 2001/18/EG. In België is het de Adviesraad voor Bioveiligheid (ARB), met de ondersteuning van externe experten en van de SBB, die deze analyse uitvoert.
Bijdragen van de Adviesraad voor Bioveiligheid en van de SBB (onderstaande figuur)
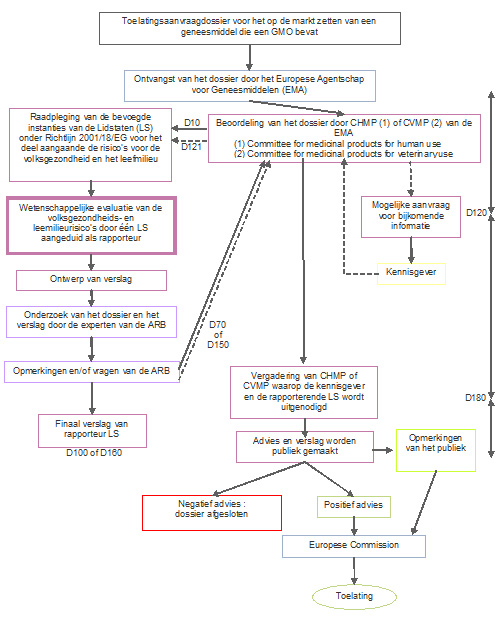
De preliminaire beoordelingsrapporten, opgesteld door de rapporteur en de co-rapporteur van de EMA en gevalideerd door één van bovenvermelde comités, bevatten een onderdeel voor de risicobeoordeling voor het leefmilieu (ERA) van het "GGO" geneesmiddel. De ERA betreft de risico's voor de biotische omgeving, alsook de eventuele risico's voor de naasten van de patiënt of het verzorgend personeel, en voor de volksgezondheid. In dit deel van het rapport worden ook de eventuele mankementen van de ERA opgelijst, en worden, indien nodig, vragen voor de kennisgever gesuggereerd.
Het "ERA" gedeelte van het rapport wordt aan de ARB overgemaakt, alsook aan alle bevoegde overheden aangeduid door ieder lidstaat, die voorafgaandelijk toegang gekregen hebben tot het "ERA" gedeelte van het dossier.
De ARB stelt een advies op over het rapport van de rapporteur van de EMA over het ERA gedeelte, en indien nodig vermeldt het de punten die de rapporteur niet voldoende in rekening genomen zou hebben, alsook vragen die het aan de kennisgever zou willen stellen. Dit advies wordt naar EMA opgestuurd, en de ARB zal telkens een advies opstellen en naar EMA opsturen, over ieder rapport van de rapporteur, opgesteld ten gevolge van de antwoorden van de kennisgever op de gestelde vragen.
Besluitvorming
Zoals eerder aangeduid, is het de Europese Commissie die de uiteindelijke beslissing neemt over het op de markt brengen van een geneesmiddel, waarvoor het dossier volgens de gecentraliseerde procedure werd ingediend.
De volledige wetenschappelijke rapporten van ieder door de Commissie toegelaten geneesmiddel, volgens de gecentraliseerde procedure en volgend op een evaluatie van het CHMP of the CVMP van EMA, zijn publiek toegankelijk op de website van EMA (EPAR – European Public Assessment Report). De EPAR bevat de bijsluiter van het product en een samenvatting van diens eigenschappen, vertaald in verschillende Europese talen, alsook de wetenschappelijke discussie en de uitgevoerde evaluatie (enkel in het Engels). De toelatingen zijn vijf jaar geldig, en de aanvragen voor hernieuwing moeten ingediend worden bij EMA ten minste zes maanden voor het verstrijken van deze geldigheidsperiode.